Signaling-network models are directed graphs: nodes are proteins and genes, edges are activations or inhibitions. Curating one network takes years of literature work. Tewari et al. (2025) tested whether a frontier LLM, given only a gene list, could reconstruct such a network. On cardiomyocyte hypertrophy, fibroblast activation, and mechanosignaling, they reported 24-58% of reactions recovered(model-dependent).
ReconBench is an Inspect benchmark that implements the paper’s tool-free protocol. It reproduces the recall figures and adds precision and F1, which the paper did not report. For Gemini 2.0, paper-reported recall was ~27% but ReconBench measures ~0%. That gap traces to a regex-extractor / output-format mismatch rather than model regression.
Table of contents
- Why this benchmark
- Task and protocol
- Pipeline
- Scoring
- Results: 10 epochs, three models
- Replication vs. paper baseline
- The Gemini extraction trap
- What does “ground truth” mean here
- Limits, and what’s next
Why this benchmark
Most LLM benchmarks stress math, code, and multiple choice. Signaling-network reconstruction is long-context structured retrieval over biology. From about $N \approx 100$ gene symbols, the model must recover the $\approx!200$ signed directed edges in a curated ground truth. Runs are single-shot with no retrieval. What counts as a valid reaction is spelled out only in natural-language instructions, not in a formal machine-readable schema.
Scoring stays clean for three reasons.
- It is verifiable. The ground truth is a published, validated reaction list. Recall and precision are well defined. There is no judge model in the loop.
- It tests latent biological knowledge. Without tools, the model must rely on pretraining, and gaps in coverage reflect gaps in the literature.
- It exposes the harness. Outputs are free-form prose, and scoring is a regex pass. Every choice in the extractor is part of what the benchmark measures.
Task and protocol
Each run covers one network. ReconBench uses the cardiomyocyte hypertrophy network from Ryall et al. (2012): 107 species (gene/protein/phenotype nodes) and the paper’s 193 reaction rows.
Those 193 rows are not the scoring denominator. ReconBench scores pairwise signed interactions, so it drops the 17 input rows, expands Boolean source rules such as A & B => C into pairwise edges, and deduplicates identical (source, target, effect) triples. For the hypertrophy sheet this yields 191 unique ground-truth edges from 195 expanded edge mentions, and that 191 is the $|G|$ used below. The task records these counts under ground_truth_accounting so the denominator is auditable rather than implied.
The prompt follows the paper’s Methods: a three-step scaffold delivered across one multi-turn conversation.

Figure 1. The three-step iterative scaffold (one run).
Step 1 dumps the full 107-node gene list into a single message. Step 2 asks for > 0 but < max_connections directed interactions among the first 20 nodes, with each effect labeled stimulated or inhibited; max_connections is the maximum out-degree of any source node in the ground truth. Step 3 repeats “do the same operation for the next 20 nodes” until the gene list is exhausted (5 continuation turns).
The scoring extractor reads every assistant turn in the transcript. Ten epochs per model.
Pipeline
The harness is small. Inspect handles batching, retries, and trajectory logging; ReconBench supplies the prompts, the regex extractor, and the scorer.

Figure 2. The ReconBench harness.
@solver
def reconbench_solver() -> Solver:
async def solve(state, generate):
chunks = state.metadata["chunks"]
await generate(state) # steps 1 and 2
for chunk in chunks[1:]: # step 3, repeated
state.messages.append(
ChatMessageUser(content=make_continuation_prompt(len(chunk)))
)
await generate(state)
return state
return solve
The extractor reads every assistant turn across the entire transcript. The iterative scaffold spreads the answer across $\lceil 107/20\rceil = 6$ turns.
Scoring
A reaction is the triple (source, target, effect) with effect ∈ {stimulated, inhibited}. Given the returned set $R$ and ground truth $G$,
$$ \text{recall} = \frac{|R \cap G|}{|G|}, \quad \text{precision} = \frac{|R \cap G|}{|R|}, \quad \text{F1} = \frac{2 \cdot \text{precision} \cdot \text{recall}}{\text{precision} + \text{recall}}. $$
The extractor handles four output shapes the paper’s models produced:
- prose:
ADRB2 stimulates RAC1 - arrow:
RAC1 -> MAP3K1(with a window of context to disambiguate sign) - pipe table:
| ADRB2 | RAC1 | stimulated | - field block:
Input Node: ADRB2 ... Affected Node: RAC1 ... Effect: stim.
The extractor checks each edge against the allowed node set, so invented symbols cannot inflate recall or precision.
Results: 10 epochs, three models
Run via OpenRouter on 2026-04-30: 10 epochs per model on the hypertrophy network, summed across runs.
| Model | Recall | Precision | F1 |
|---|---|---|---|
| Claude 3.7 Sonnet | 57.2% | 50.5% | 53.7% |
| GPT-4.1 | 24.4% | 29.5% | 26.7% |
| Gemini 2.0 Flash | 0.2% | 44.4% | 0.4% |
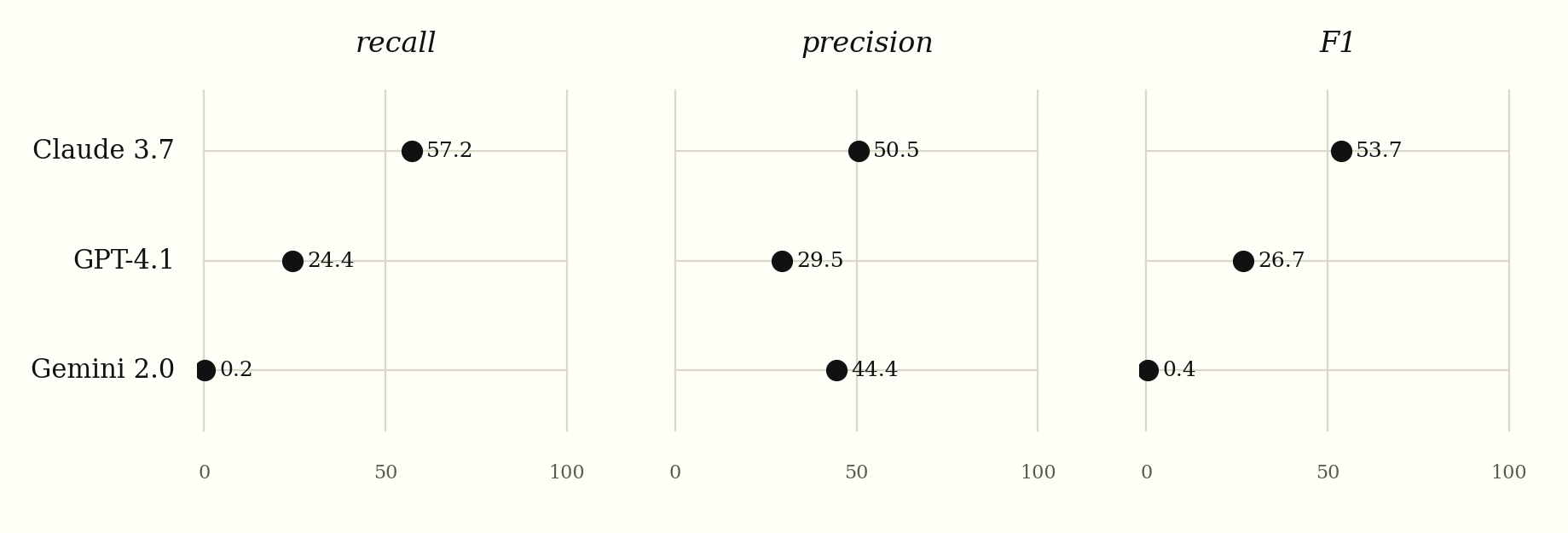
Figure 3. Reconstruction of the cardiomyocyte hypertrophy network (10 epochs).
Claude 3.7’s precision and recall track each other. They sit within seven points (50.5% and 57.2%), so recall does not come from a flood of low-confidence guesses. F1 of 53.7% is the highest reported for a tool-free LLM on this network in the literature consulted (Tewari et al. and its references).
GPT-4.1 commits to fewer edges, but the ones it commits to land more often than not. Gemini’s near-zero recall paired with 44% precision points to an extraction problem: the extractor parsed only a few Gemini edges, but several were correct.
Replication vs. paper baseline
The paper does not publish per-network, per-model recall as standalone numbers; it reports a band of 26.70-58.12% across all three networks and three models. Comparing against the band’s anchor points, and tracing each model’s recall through the harness:
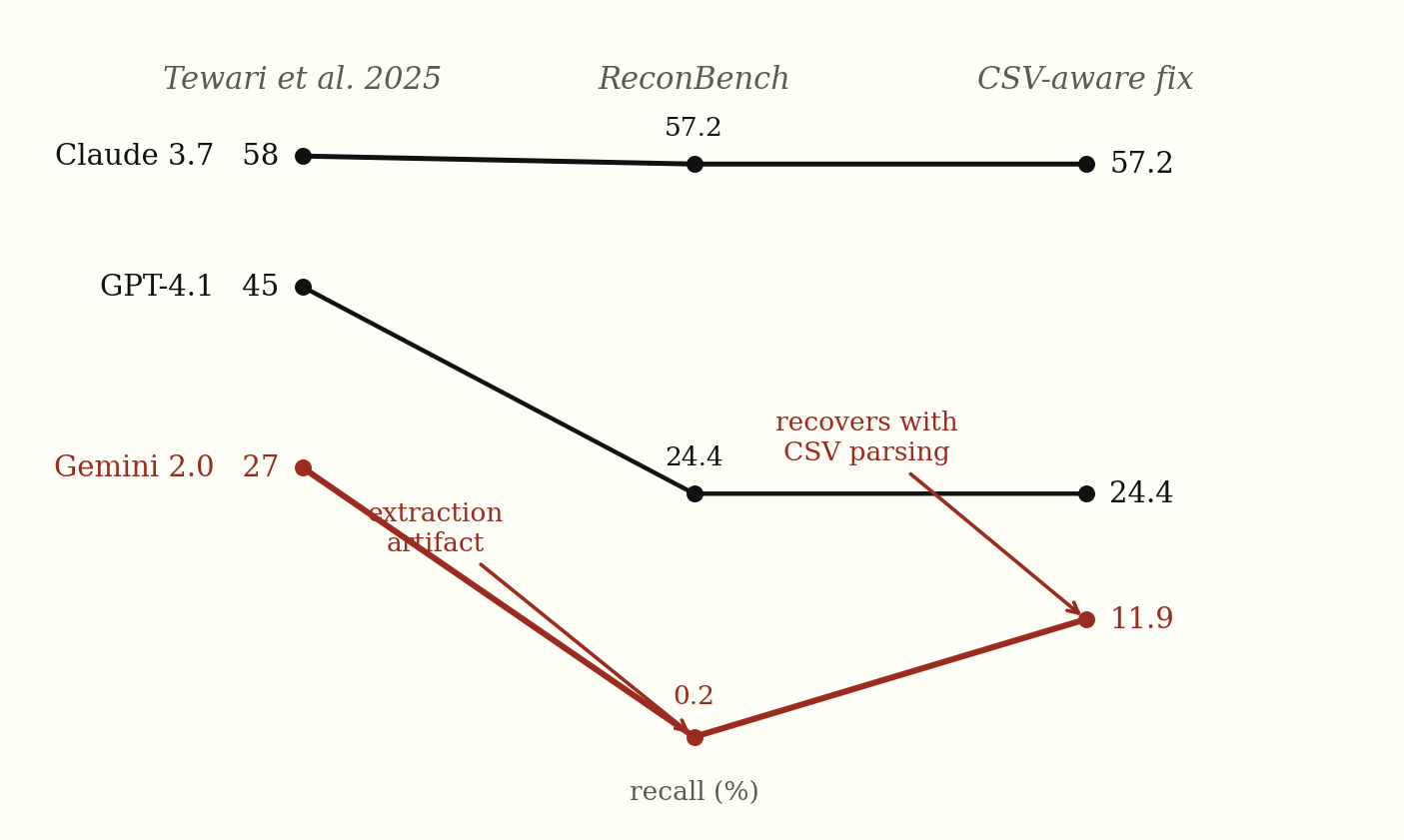
Figure 4. Recall across three stages: paper-reported, ReconBench’s raw extractor, and after the CSV-aware fix. GPT-4.1 drops between paper and ReconBench and stays there — a genuine shortfall. Gemini collapses to 0.2% then recovers to 11.9%, because only it emitted bare CSV; that dip was an extraction artifact, not a reasoning gap (next section). Claude holds throughout.
| Model | Paper recall (~) | ReconBench recall |
|---|---|---|
| Claude 3.7 Sonnet | 58% | 57.2% ✓ |
| GPT-4.1 | 45% | 24.4% |
| Gemini 2.0 Flash | 27% | 11.9%* |
*Gemini’s recall after correcting the scoring harness; the initial run measured 0.2%. The next section explains why.
Claude’s results match the paper. GPT-4.1 underperforms by ~20 points. Gemini lands at 11.9% — but only after a correction to the scoring harness, because the initial run measured 0.2%. That collapse turned out to be an artifact of the harness rather than the model, which is worth walking through on its own.
The Gemini extraction trap
Reading Gemini 2.0’s transcripts via Inspect Scout shows the model emits CSV tuples:
ADRB2, RAC1, Stimulated
ADRB2, ADCY, Stimulated
RAC1, MAP3K1, Stimulated
The paper’s extractor, and the one used here, aligned to it, keyed on prose verbs (stimulates, inhibits) and arrows (->, =|). The regex ignored bare comma-separated triples, so the harness dropped valid edges whenever the model returned CSV. The model can be correct while the harness is blind.

Figure 5. What the regex catches, and what each model emits.
The right response is methodological: fix the evaluation protocol rather than patch it only for Gemini. A tool-free reconstruction benchmark should use either:
- a structured-output constraint (JSON schema, tool call), or
- an extractor that supports a documented set of shapes and reports the observed shape distribution.
That fix came after the fact. Re-scoring the same 10-run Gemini log set with a CSV-aware extractor confirms the gap was an extraction bug rather than a reasoning one: Gemini’s measured recall rises from 0.2% to 11.9% (the recovery in the final column of Figure 4), precision from 44.4% to 55.3%, and F1 from 0.4% to 19.6%. Phase 2 goes further, adding a structured JSON-output mode so the next comparison does not depend on free-form regex coverage at all.
What does “ground truth” mean here
The Ryall network is a manually curated, validated subset of cardiomyocyte signaling rather than the full underlying system. An edge that appears in a model’s output and not in $G$ counts as a false positive even when it is biologically plausible: the LLM might be right and the curators silent. This is a known limitation of recall/precision metrics against curated graphs, and it is one reason the paper’s “validation accuracy” experiments, which check model-predicted responses to perturbations against held-out wet-lab data, are a more demanding test than reaction-set overlap.
ReconBench Phase 1 stops at structural reconstruction. Read 50% precision as half of emitted edges matching the curated list; that says nothing about biological truth outside that list.
Limits, and what’s next
ReconBench Phase 1 is narrow:
- One network: hypertrophy only. Adding fibroblast and mechanosignaling reproduces the rest of Tewari et al.’s grid but does not change the methodology.
- No tools: Phase 2 asks whether retrieval closes the gap.
- Free-form output: as discussed, this conflates capability and format.
Phase 2 (tool-augmented agents): give the agent KEGG, STRING, and Reactome lookups, plus a structured-output channel. Measure how much retrieval closes the recall gap, and at what precision cost.
Phase 3 (predictive validation): move from structure to dynamics. Translate LLM-generated networks into Netflux logic-based ODEs (the paper does this) and score them on perturbation responses. This is a stricter test: a network can have high recall and still mispredict because it is missing a single bottleneck edge.
The harness, data, and notebooks are in reconbench/, with the Inspect Scout walkthrough at notebooks/scout_analysis.ipynb.
Citation
Amanfu, Robert. "ReconBench: Benchmarking LLMs on Cardiac Signaling Network
Reconstruction." Robert's Bytes (May 2026).
https://pubs.onrender.com/posts/reconbench/
References
- Tewari, J., Dahl, B. W., Saucerman, J. J. (2025). Benchmarking of signaling networks generated by large language models. bioRxiv 2025.07.28.667217. link
- Ryall, K. A., Holewinski, R. J., Van Eyk, J. E., Saucerman, J. J. (2012). Network reconstruction and systems analysis of cardiac myocyte hypertrophy signaling. J. Biol. Chem.
- UK AI Security Institute. Inspect: a framework for evaluating language models. inspect.aisi.org.uk
- Meridian Labs. Inspect Scout. meridianlabs-ai.github.io/inspect_scout