This is a living document and will change over time. The code and data will be open-sourced
Introduction
A problem in two acts
Heart failure, a condition characterized by insufficient cardiac output to meet the body’s needs1, affects approximately 5 million Americans2. This disease accounts for an estimated 20% of all hospital admissions for persons over 652, marking it a major health concern.
Heart failure can result from various diseases including myocardial infarction, coronary artery disease (ischemic cardiomyopathy), and genetic factors (hypertrophic cardiomyopathy). Furthermore, idiopathic cardiomyopathy accounts for a considerable number of cases where the cause remains unknown. The symptoms across these conditions are often fatigue, breathlessness, and exercise intolerance1. One shared clinical observation is high levels of catecholamines, hormones that stimulate the heart. The elevated levels is known to impact various signaling pathways3.
The β1-adrenergic receptor pathway, crucial for how hard the heart pumps, is one of the signaling pathways affected. Chronic exposure to catecholamines can desensitize this pathway, leading to a loss of responsiveness and reduced cardiac output4. Notably, altered protein levels of signaling components in the pathway, especially the downregulation of the β1-adrenergic receptor by 50%5, have been observed. Other components such as the sarcoplasmic reticulum Ca2+ ATPase (SERCA)6, G-protein receptor kinase (GRK2)7, and phospholamban8 are also affected.
Identifying the most influential changes leading to heart failure is complex9. Previous studies have focused on a limited number of pathway perturbations or studied them in isolation. There is a need for a comprehensive approach to understand how these changes contribute to heart failure and design therapies to restore signaling pathway responsiveness. Current therapies, such as certain β-blockers10 and the left ventricular assist device (LVAD)11, aim to do just that.
Simulation as a tool
Computational modeling of the heart has been vital for understanding heart physiology and pathology12. Such models can effectively illustrate how genes impact organ function in human populations13. Britton et al. used population computational models to study human variability in electrophysiological activity by randomly altering model parameters14. Similarly, Walmsley et al. developed population models of normal and failing cardiac myocytes by changing parameter values associated with significantly altered genes in heart failure15. However, both studies relied on idealized probability distributions, not actual gene expression levels.
A data-driven approach combining compuational models with RNAseq and microarray data could prove useful. Even with limited patient-specific post-transcriptional and post-translation regulation data, we hypothesize that mRNA expression integration would suffice to predict critical aspects of different heart failure etiologies.
By mapping differential gene expression in heart failure patients onto protein species in our model16, we’ve studied alterations comprehensively. Sensitivity analysis has enabled us to predict genes that contribute or compensate for the loss of adrenergic responsiveness in human heart failure. Importantly, distinct patient model responses have emerged from diverse patient transcriptional profiles, demonstrating correlations with clinical measures.
Materials and Methods
Computational model of β1-adrenergic signaling and EC coupling in rabbit mouse ventricular myocytes
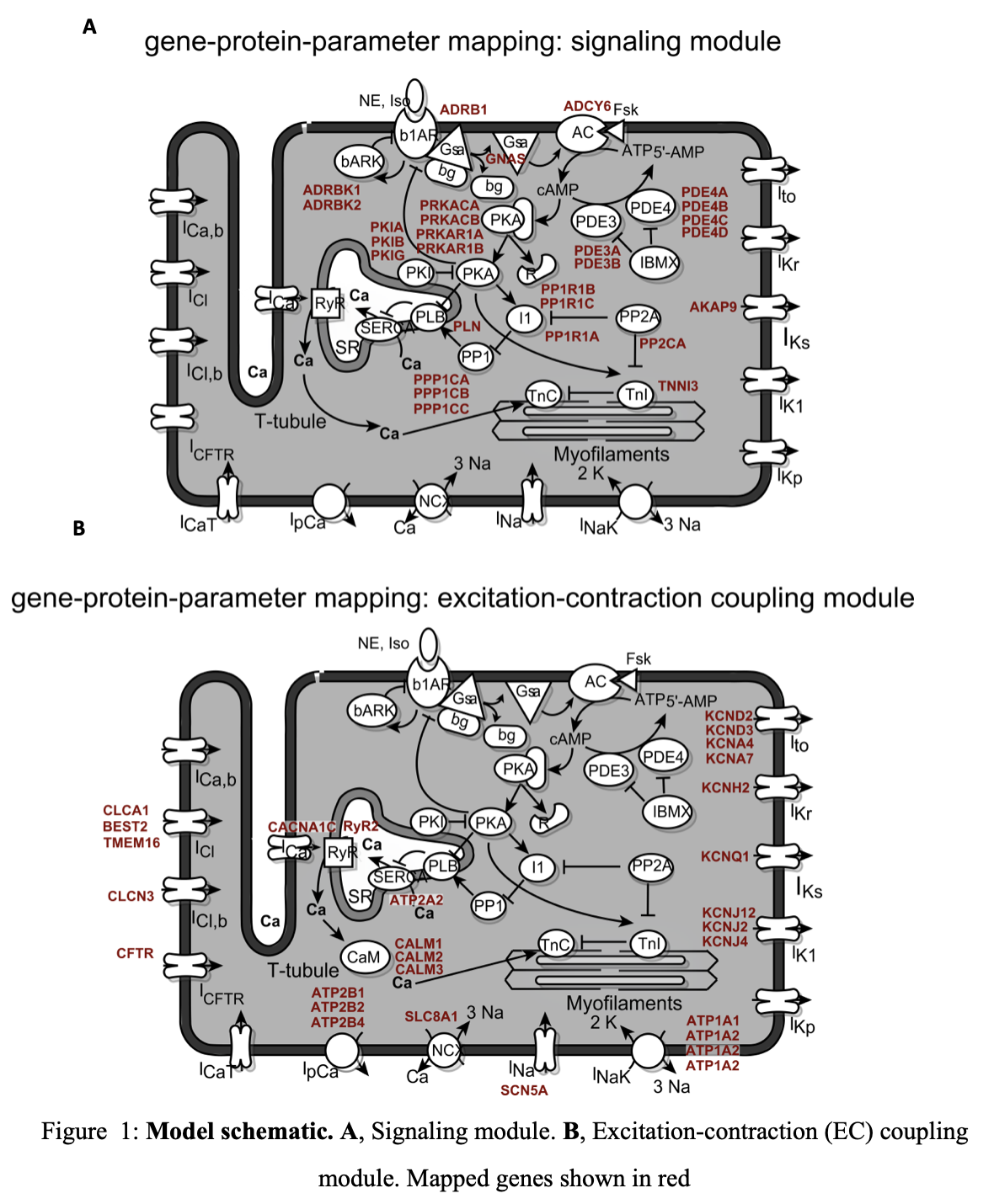 We integrated experimentally determined mRNA expression profiles with a previously developed model of the rabbit ventricular myocyte, which includes EC coupling, CaMKII and PKA signaling16 (Figure 1). Computational modeling was performed in MATLAB (The Mathworks, Natick MA).
We integrated experimentally determined mRNA expression profiles with a previously developed model of the rabbit ventricular myocyte, which includes EC coupling, CaMKII and PKA signaling16 (Figure 1). Computational modeling was performed in MATLAB (The Mathworks, Natick MA).
Gene-protein parameter mapping
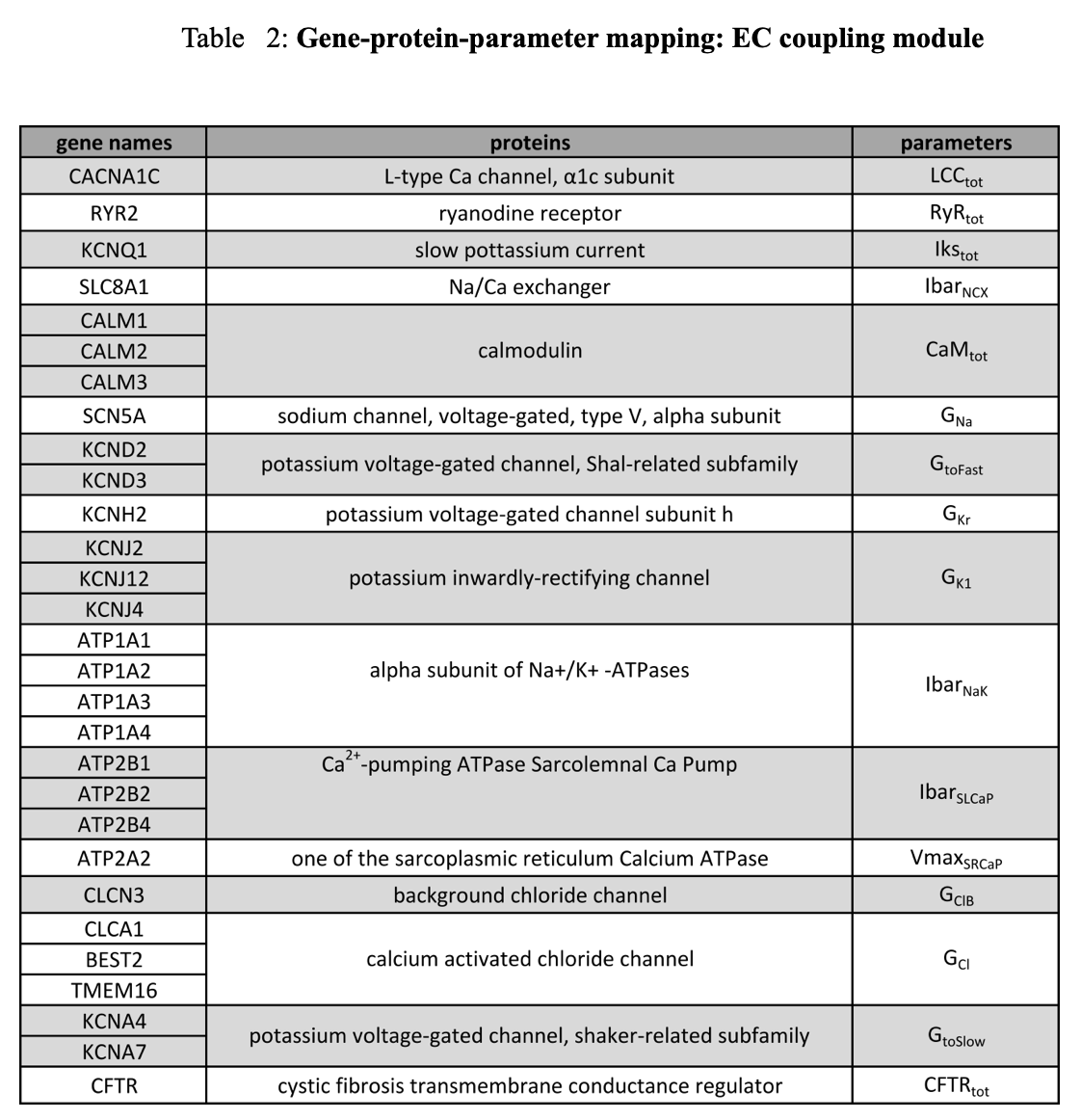
Gene-protein-parameter mapping was done for 59 genes associated with 30 proteins, and was mapped to 30 parameters in the model (Table 1 & 2). Specific parameter/gene/symbol assignments were made using the NCBI gene17, supplemented by literature references for chloride channels17.
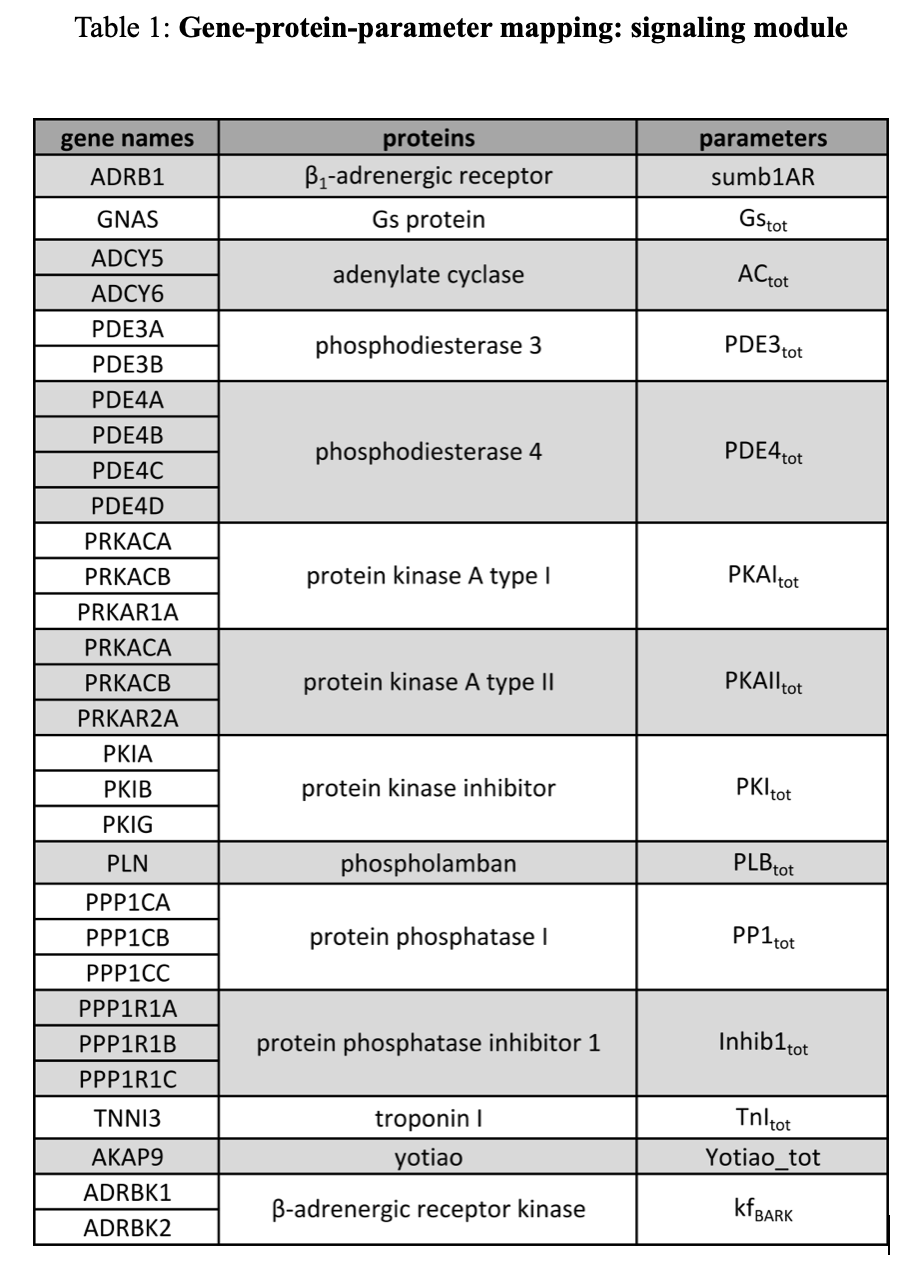 Changes in ion channel gene expression were mapped by altering corresponding ion channel conductance parameters. The activity of the β-adrenergic kinase was altered by modifying the phosphorylation rate. Other gene expression levels were mapped to total protein concentration parameters.
Changes in ion channel gene expression were mapped by altering corresponding ion channel conductance parameters. The activity of the β-adrenergic kinase was altered by modifying the phosphorylation rate. Other gene expression levels were mapped to total protein concentration parameters.
 The fold change when a single gene’s expression (G) is mapped to a parameter was determined as:
The fold change when a single gene’s expression (G) is mapped to a parameter was determined as:
$$FCHF_{\text{condition}} = \frac{G_{\text{HF condition}}}{G_{\text{normal}}}$$
When multiple genes are mapped to a given parameter, fold change was determined as:
$$FCHF_{\text{condition}} = \frac{G1_{\text{HF condition}} + G2_{\text{HF condition}} + \ldots + Gn_{\text{HF condition}}}{G1_{\text{normal}} + G2_{\text{normal}} + \ldots + Gn_{\text{normal}}} $$
Parameters were modified by multiplying the normal parameters by the fold change. For the mRNA models, this assumes a linear relationship between mRNA expression level, protein abundance, and the corresponding parameter value.
Gene expression data
Gene expression data was obtained from the cardioGenomics consortium18. The database contains gene expression profiles of myocardial samples from patients undergoing cardiac transplantation with heart failure arising from different etiologies: idiopathic dilated (27 patients), ischemic (31 patients), and hypertrophic (5 patients) cardiomyopathies. The database also includes profiles of “normal” (14 patients) organ donors whose hearts were deemed unsuitable for transplantation. Gene expression data was analyzed using MATLAB’s Bioinformatics Toolbox.
Simulation conditions
All simulations were first run to steady state for 30 seconds, and Ca2+ current and action potential were simulated. Six functional readouts were quantified: diastolic Ca2+ transient level (Camin), Ca2+ transient amplitude (Caamp), time for 80% decay of Ca2+ transient (CaT80), resting membrane potential (RMP), action potential duration at 50% repolarization (APD50), and action potential duration at 70% repolarization (APD70).
Ca2+ transients and action potential were simulated at 1 Hz pacing. For patient simulations, fold change for each patient (normal, idiopathic, ischemic and hypertrophic) was calculated with reference to the normal population mean. Each patient model was run to steady state for 30 seconds. For simulations with isoproterenol (ISO), a dose of 1 μM was used.
Sensitivity analysis
Sensitivity analysis was performed by perturbing a single model parameter with the corresponding fold change in mRNA expression. The effect of a single-gene perturbation of the ith gene on a functional readout (FR) is quantified (similar to Feilun et al.; in preparation) by:
$$ Z_{FRi} = \frac{FRi - FR_{\text{normal}}}{FR_{\text{HF condition}} - FR_{\text{normal}}} $$
Statistical analysis
A Pearson correlation matrix was determined by finding correlation between each model output and clinical measures. A coefficient was considered significant for p <0.1 without corrections for multiple comparisons.
Results
Validating computational models of human heart failure
Despite limited correlation between protein and mRNA expression, we hypothesized that mRNA expression measured by microarrays would be sufficient to predict the loss of adrenergic responsiveness seen in heart failure. To test this, transcriptional profiles of heart failure patients with different etiologies was integrated with a previously developed EC coupling and β1- adrenergic signaling model (Tables 1 & 2).
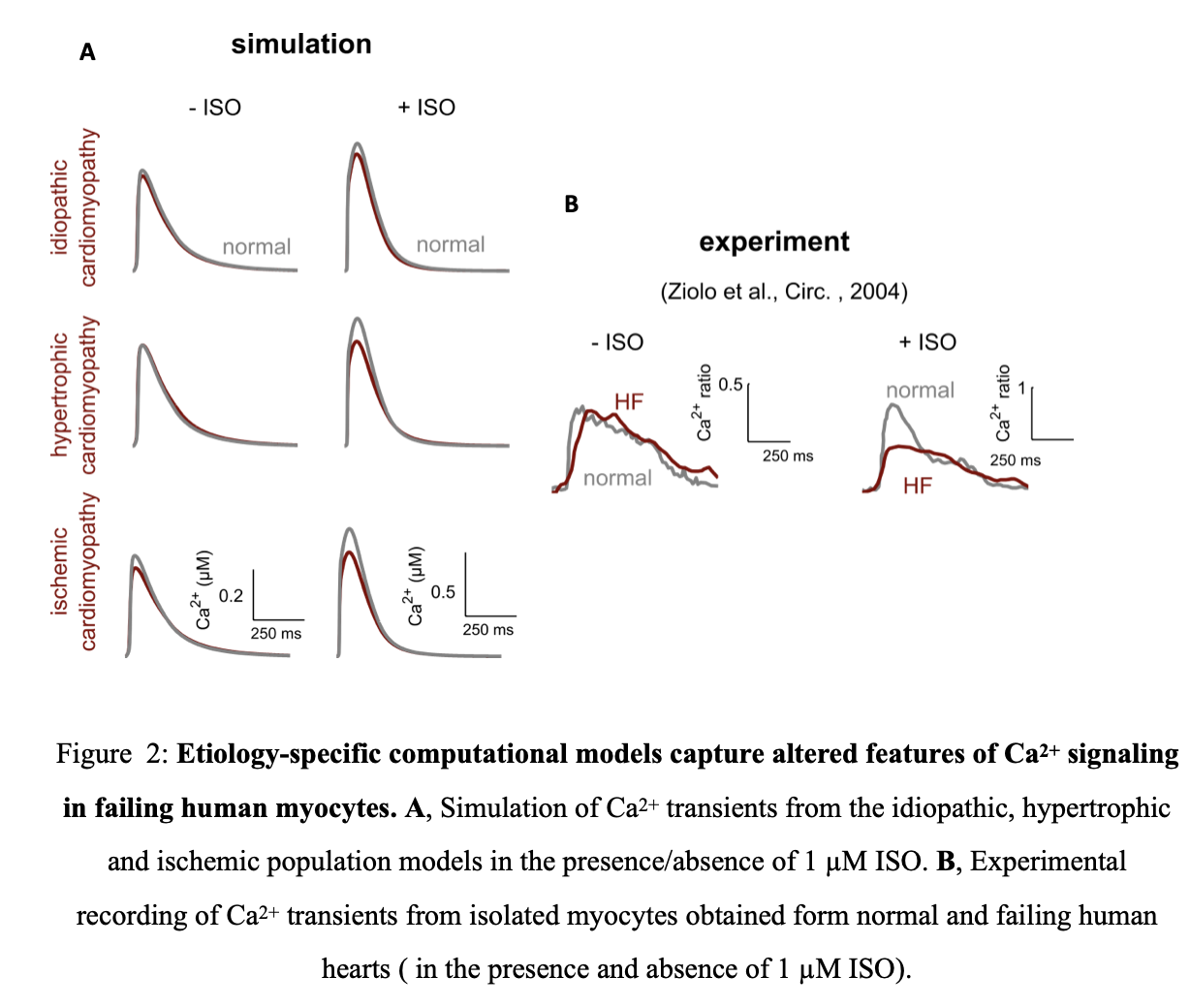 The models of idiopathic, hypertrophic and ischemic cardiomyopathies was able to capture essential qualitative features of altered Ca2+ signaling in heart failure including reduced adrenergic responsiveness seen in heart failure (Figure 2A).
The models of idiopathic, hypertrophic and ischemic cardiomyopathies was able to capture essential qualitative features of altered Ca2+ signaling in heart failure including reduced adrenergic responsiveness seen in heart failure (Figure 2A).
Experimentally, this manifests as lowered Ca2+ amplitude in heart failure patients compared to normal patients (Figure 2 B). Although model simulations differ quantitatively from the experimental measurement, Ziolo et al.19 measured Ca2+ transients from a single failing human heart while our model includes information from a large patient population. Thus the quantitative differences could be due to patient transcriptional profile diversity. The dearth and variability of single cell Ca2+ measurements of isolated human ventricular myocytes makes it difficult to verify this prediction.
Single gene-protein-parameter perturbations identify therapeutic targets for restoring adrenergic responsiveness
Single gene-protein-parameter perturbations were performed to examine the contribution of individual genes to the overall changes in the 6 functional readouts. For each simulation, only one parameter was altered from normal.
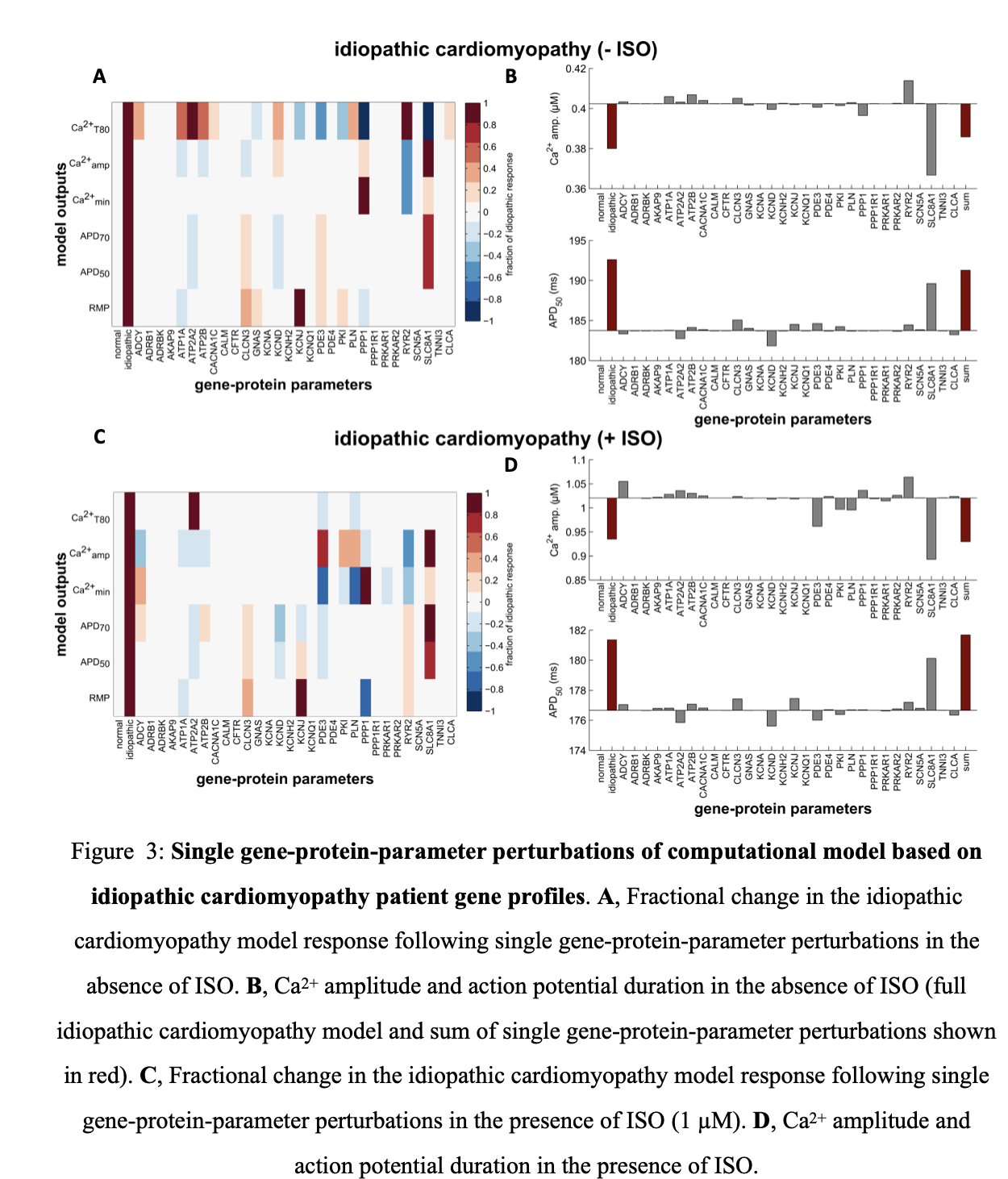 When parameter mapping was based on average gene profiles of idiopathic patients, the Na-Ca exchanger (SLC8A1), which is upregulated in this condition, had the largest effect on both Ca2+ transient and action potential readouts with and without ISO stimulations( Figure 3 A,C). Inclusion of this parameter reduced Ca2+ amplitude and increased APD50, consistent with observed changes seen in heart failure. The model however captures compensatory mechanisms that restore adrenergic responsiveness including ryanodine receptor (RYR2) and adenylyl cyclase (ADCY) which are both upregulated (Figure 3 B, D).
When parameter mapping was based on average gene profiles of idiopathic patients, the Na-Ca exchanger (SLC8A1), which is upregulated in this condition, had the largest effect on both Ca2+ transient and action potential readouts with and without ISO stimulations( Figure 3 A,C). Inclusion of this parameter reduced Ca2+ amplitude and increased APD50, consistent with observed changes seen in heart failure. The model however captures compensatory mechanisms that restore adrenergic responsiveness including ryanodine receptor (RYR2) and adenylyl cyclase (ADCY) which are both upregulated (Figure 3 B, D).
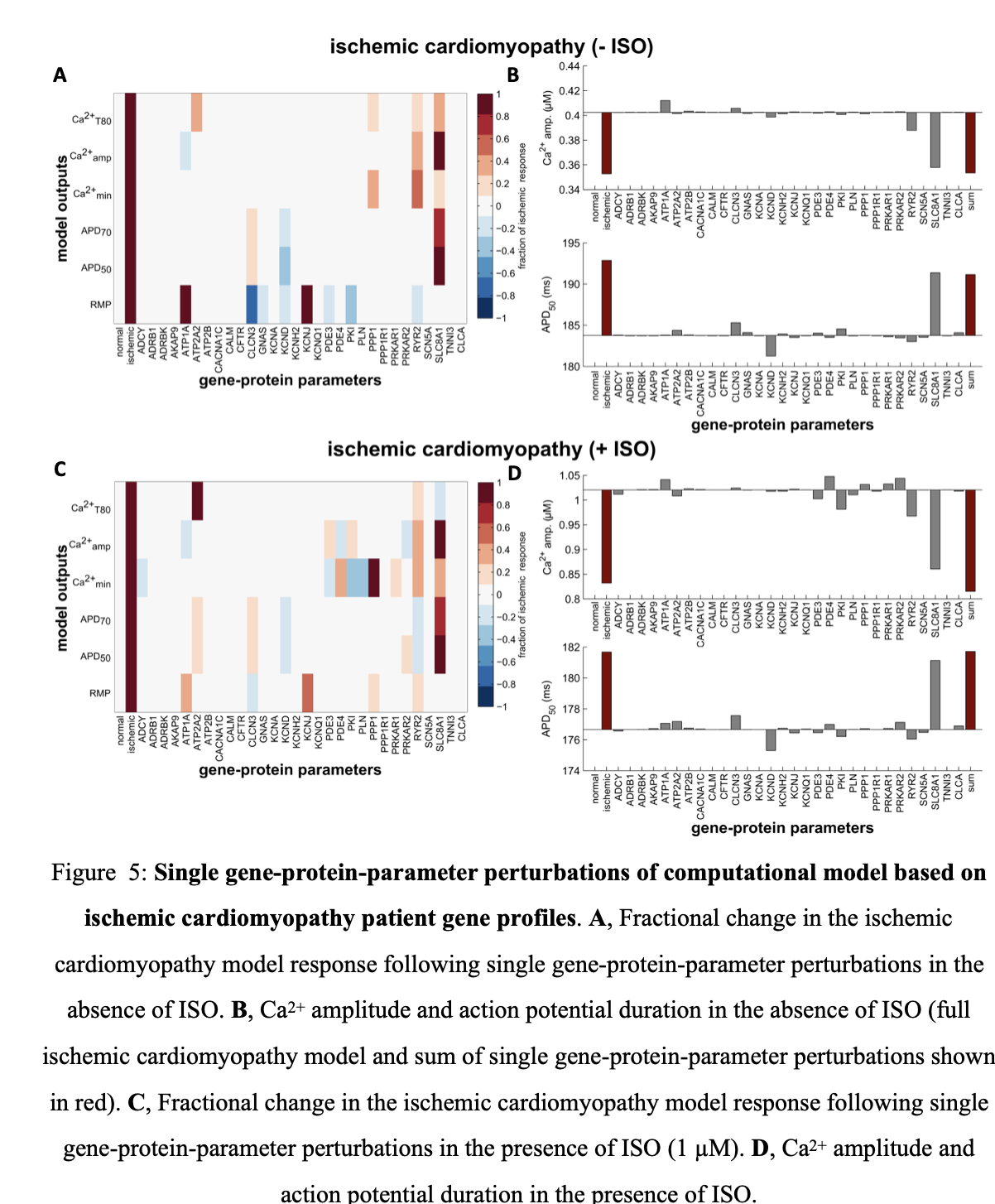
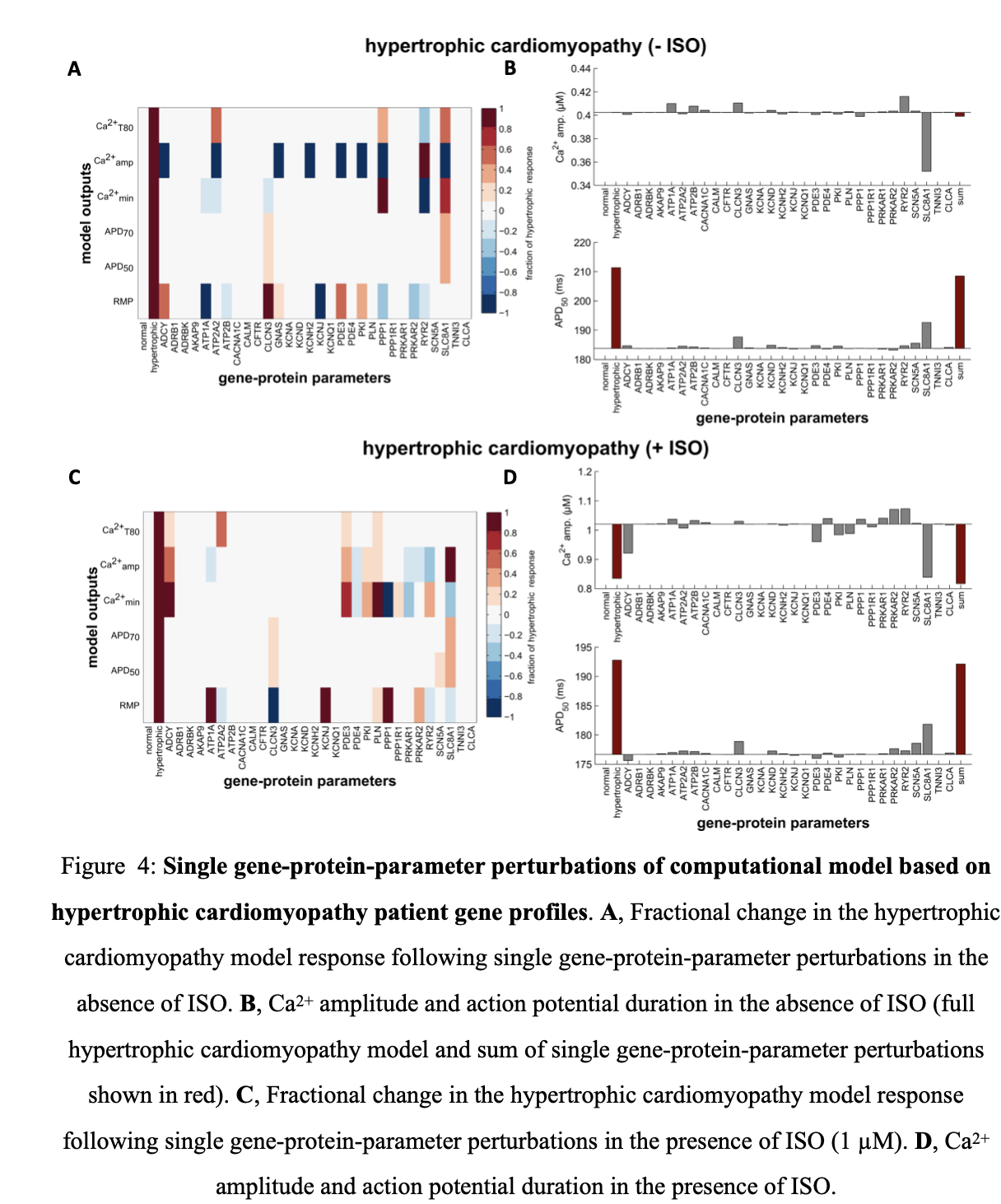 A similar trend is seen in model simulations with parameters altered using expression profiles of patients with hypertrophic cardiomyopathy (Figure 4). The Na-Ca exchanger still has the largest effect but different compensatory changes, which enhance adrenergic responsiveness, are present in this condition. This includes PKA type II (PRKAR2, upregulated) and the inward rectifying potassium channel (KCNH2, downregulated). For model simulations with parameters altered using gene profiles for ischemic cardiomyopathy patients, deleterious changes include the slow transient outward potassium channel (KCNA, upregulated). However, similar to the other two cardiomyopathies, Na-Ca exchanger expression is the major driver of altered Ca2+ transients and electrophysiology (Figure 5).
A similar trend is seen in model simulations with parameters altered using expression profiles of patients with hypertrophic cardiomyopathy (Figure 4). The Na-Ca exchanger still has the largest effect but different compensatory changes, which enhance adrenergic responsiveness, are present in this condition. This includes PKA type II (PRKAR2, upregulated) and the inward rectifying potassium channel (KCNH2, downregulated). For model simulations with parameters altered using gene profiles for ischemic cardiomyopathy patients, deleterious changes include the slow transient outward potassium channel (KCNA, upregulated). However, similar to the other two cardiomyopathies, Na-Ca exchanger expression is the major driver of altered Ca2+ transients and electrophysiology (Figure 5).
Model outputs predict patient clinical measures
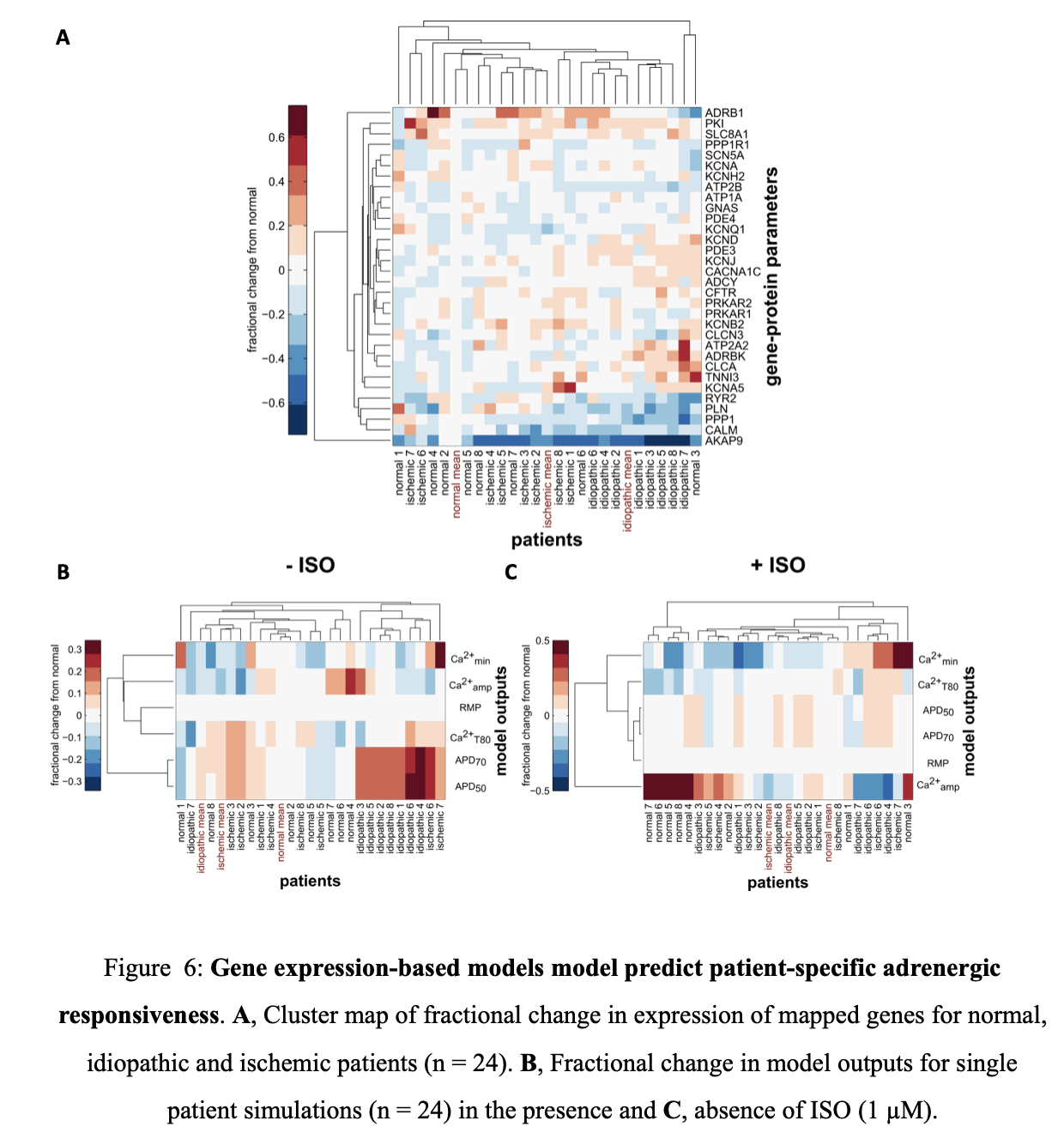 Clustering of mapped genes for normal, idiopathic and ischemic patients was done to better understand the influence of individual patient gene profiles on adrenergic responsiveness (Figure 6 A). Clustering identified idiopathic patients with reduced expression of various signaling components including protein phosphatase I (PPP1), calmodulin (CALM) and the A-kinase anchoring protein yotiao (AKAP9). There was significant patient variability in other parameters, including the Na-Ca exchanger (SLC8A1), the major driver in the population averaged gene profiles. Clustering of individual patient profiles without ISO stimulation suggests that idiopathic patients have significant altered electrophysiology with prolonged action potential duration.
Clustering of mapped genes for normal, idiopathic and ischemic patients was done to better understand the influence of individual patient gene profiles on adrenergic responsiveness (Figure 6 A). Clustering identified idiopathic patients with reduced expression of various signaling components including protein phosphatase I (PPP1), calmodulin (CALM) and the A-kinase anchoring protein yotiao (AKAP9). There was significant patient variability in other parameters, including the Na-Ca exchanger (SLC8A1), the major driver in the population averaged gene profiles. Clustering of individual patient profiles without ISO stimulation suggests that idiopathic patients have significant altered electrophysiology with prolonged action potential duration.
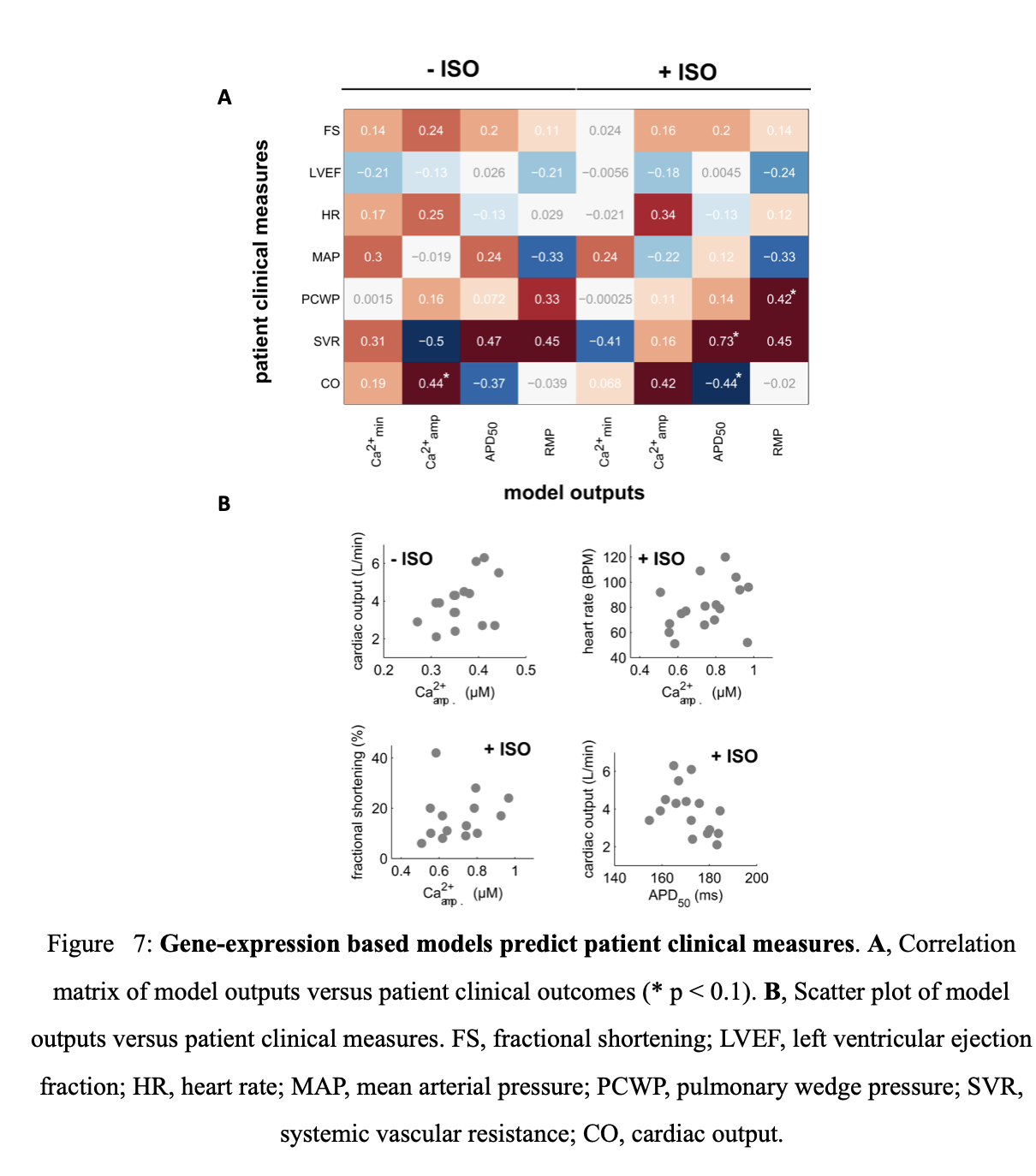 Following ISO stimulation, idiopathic and ischemic cardiomyopathy patients predominantly have reduced adrenergic responsiveness although there is variability in diastolic Ca2+ levels (Ca2+min). This highlights the diversity present in heart failure gene profiles and presents a significant challenge in developing appropriate therapeutic interventions. Model driven drug discovery can be useful in this endeavor especially if model outputs are predictive of clinical measures. Regression analysis was performed to determine which patient clinical measures are adequately predicted by the model’s functional readouts (Figure 7 A, G). Ca2+ amplitude without ISO stimulation was positively correlated with cardiac output (p = 0.09). Model outputs are thus mildly predictive of particular clinical measures.
Following ISO stimulation, idiopathic and ischemic cardiomyopathy patients predominantly have reduced adrenergic responsiveness although there is variability in diastolic Ca2+ levels (Ca2+min). This highlights the diversity present in heart failure gene profiles and presents a significant challenge in developing appropriate therapeutic interventions. Model driven drug discovery can be useful in this endeavor especially if model outputs are predictive of clinical measures. Regression analysis was performed to determine which patient clinical measures are adequately predicted by the model’s functional readouts (Figure 7 A, G). Ca2+ amplitude without ISO stimulation was positively correlated with cardiac output (p = 0.09). Model outputs are thus mildly predictive of particular clinical measures.
Discussion
In this study, high-throughput measurement of human mRNA expression was integrated into a mechanistic model of cardiac myocyte electrophysiology and β1-adrenergic signaling. Model predictions on Ca2+ dynamics in failing human hearts was validated against an independent experiment19 and sensitivity analysis identified the Na-Ca exchanger (NCX) as a major contributor to altered Ca2+ transient and action potential duration in all the heart failure etiologies tested. Simulations with individual patient transcriptional profiles resulted in distinct patient model Ca2+ responses that were correlated with cardiac output.
Genes contributing to altered β1-adrenergic signaling and EC coupling
Patients with heart failure have depressed ventricular contractility20, also seen in ventricular myocytes isolated from heart failure patients21. These contractile deficits are associated with altered Ca2+ transient and action potentials, consistent with our model simulations22. Yet experimental data investigating the molecular mechanisms underlying these deficits are limited and inconsistent.
For example, experimental measurement of protein levels of SERCA show disagreement in the literature. SERCA, a Ca2+ ATPase that resides in the sarcoplasmic reticulum, is responsible for Ca2+ removal from the cytosol. SERCA down regulation is thought to underlie the slow decay rate of Ca2+ transient in the failing heart23. However, some direct protein measurements have shown no change in SERCA expression[^24,^25]. This suggests that the altered features of Ca2+ handling in the failing heart are not always caused by reduction in SERCA24.
Computational models can relate patient transcriptional profiles to clinical measures
Previous studies have used mechanistic models of cardiac myocytes to understand the genotype-phenotype gap in heart pathology. Several models have incorporated gene mutation or changes in protein expression, predicting mechanistic links to pathology[^29,^30]. Regression analysis and global sensitivity of model parameters has also guided studies in phenotypic variation[^14,^31].
While those studies simultaneously varied all parameters, results of our sensitivity analysis suggest that the heart failure phenotype can be explained by a combination of single gene interactions. This potentially reduces the search space for novel therapeutic targets. While a recent study has mapped genes to parameters in a cardiac computational model, parameter values were obtained from idealized probability distributions instead of directly incorporating measured changes in mRNA expression15. Also, our approach allows direct extension to other patient populations where transcriptional profiles have been measured including heart failure patients undergoing treatment with LVAD and β-blockers
Limitations and Considerations
There are several limitations to the overall approach developed here. This includes the use of mRNA expression which is only partially correlated with protein expression. Comparison of the mouse cardiac proteome by mass spectrometry with Affymetrix microarray showed lower correlation between the transcriptome and proteome25. While proteomic information might be preferred for integration into mechanistic computational models, the dearth of such data leaves mRNA profiles as the best available alternative. In addition, the limited number of studies measuring Ca2+ and electrophysiology of isolated human myocytes made exhaustive model validation difficult. Despite these issues, this current study indicates that mRNA profiles may be sufficient to predict patient phenotypes. A hypothesis suggested by our studies is that adrenergic responsiveness can be restored via therapeutic targeting of particular proteins. This could be tested by applying the same approach used here to datasets from patients undergoing treatment with LVAD and β-blockers. These treatments have been shown to restore adrenergic responsiveness and could confirm the targets suggested in this work. In addition, alterations in other signaling pathways besides the β1- adrenergic pathway could be studied to identify other potential therapeutic targets. Here we integrated transcriptional profiles with a rabbit computational model which shares many features of human excitation-contraction coupling26. Although no conflict arose between mappings of human genotypes to proteins in a rabbit computational model, this could be a potential issue as more comprehensive “omic” datasets are made available. In the future, our approach can be extended with the use of recently developed human ventricular myocyte models.
Conclusions
Intergration of “omic” data into mechanistic models complements standard bioinformatics approaches that search for significantly correlated genes. Such an approach can provide a predictive and mechanistic linkage between genotype and phenotype in human populations. In this study, human transcriptional profiles were integrated with mechanistic computational models of β1-adrenergic signaling and EC coupling. Integration of mRNA expression alone was sufficient to predict key aspects of the heart failure phenotype for different etiologies.
Citation
Cited as:
Amanfu, Robert. (Jun 2023). Integration of human transcriptome and cardiac systems models. Robert’s Bytes. https://pubs.onrender.com/posts/personalized_models/.
Or
@article{amanfu2023cardiacmodels,
title = "Integration of human transcriptome and cardiac systems models",
author = "Amanfu, Robert",
journal = "www.ramanfu.online",
year = "2023",
month = "Jun",
url = "https://pubs.onrender.com/posts/personalized_models/"
}
-
(Mudd, J. O. & Kass, D. A. Tackling heart failure in the twenty-first century. Nature 451, 919–928 (2008)) ↩︎ ↩︎
-
(Jessup, M. & Brozena, S. Heart Failure. N Engl J Med 348, 2007–2018 (2003)) ↩︎ ↩︎
-
(Lohse, M. J., Engelhardt, S. & Eschenhagen, T. What Is the Role of {beta}-Adrenergic Signaling in Heart Failure? Circ Res 93, 896–906 (2003)) ↩︎
-
(Ungerer, M. et al. Expression of beta-arrestins and beta-adrenergic receptor kinases in the failing human heart. Circ Res 74, 206–213 (1994)) ↩︎
-
(Bristow, M. et al. Decreased catecholamine sensitivity and β-adrenergic receptor density in failing human hearts. N Engl J Med 307, 205–211 (1982)) ↩︎
-
(Kubo, H., Margulies, K. B., Piacentino, V., 3rd, Gaughan, J. P. & Houser, S. R. Patients with end- stage congestive heart failure treated with beta-adrenergic receptor antagonists have improved ventricular myocyte calcium regulatory protein abundance. Circulation 104, 1012–1018 (2001)) ↩︎
-
(Iaccarino, G. et al. Elevated myocardial and lymphocyte GRK2 expression and activity in human heart failure. Eur. Heart J. 26, 1752–1758 (2005)) ↩︎
-
(Meyer, M. et al. Alterations of sarcoplasmic reticulum proteins in failing human dilated cardiomyopathy. Circulation 92, 778–784 (1995)) ↩︎
-
(Lou, Q., Janardhan, A. & Efimov, I. R. Remodeling of calcium handling in human heart failure. Adv. Exp. Med. Biol. 740, 1145–1174 (2012)) ↩︎
-
(Engelmeier, R. S. et al. Improvement in symptoms and exercise tolerance by metoprolol in patients with dilated cardiomyopathy: a double-blind, randomized, placebo-controlled trial. Circulation 72, 536–546 (1985)) ↩︎
-
(Ogletree-Hughes, M. L. et al. Mechanical unloading restores beta-adrenergic responsiveness and reverses receptor downregulation in the failing human heart. Circulation 104, 881–886 (2001)) ↩︎
-
(Amanfu, R. K. & Saucerman, J. J. Cardiac models in drug discovery and development: a review. Crit. Rev. Biomed. Eng. 39, 379–395 (2011)) ↩︎
-
(Gjuvsland, A. B., Vik, J. O., Beard, D. A., Hunter, P. J. & Omholt, S. W. Bridging the genotype-phenotype gap: what does it take? J. Physiol. 591, 2055–2066 (2013)) ↩︎
-
(Britton, O. J. et al. Experimentally calibrated population of models predicts and explains intersubject variability in cardiac cellular electrophysiology. Proc. Natl. Acad. Sci. U. S. A. 110, E2098–E2105 (2013)) ↩︎
-
(Walmsley, J. et al. mRNA Expression Levels in Failing Human Hearts Predict Cellular Electrophysiological Remodeling: A Population-Based Simulation Study. PLoS ONE 8, e56359 (2013)) ↩︎ ↩︎
-
(Soltis, A. R. & Saucerman, J. J. Synergy between CaMKII substrates and β-adrenergic signaling in regulation of cardiac myocyte Ca(2+) handling. Biophys. J. 99, 2038–2047 (2010)) ↩︎ ↩︎
-
(Duan, D. Phenomics of cardiac chloride channels: the systematic study of chloride channel function in the heart. J. Physiol. 587, 2163–2177 (2009)) ↩︎ ↩︎
-
(CardioGenomics: Homepage. at http://cardiogenomics.med.harvard.edu/home) ↩︎
-
(Ziolo, M. T. et al. Myocyte Nitric Oxide Synthase 2 Contributes to Blunted β-Adrenergic Response in Failing Human Hearts by Decreasing Ca2+ Transients. Circulation 109, 1886–1891 (2004)) ↩︎ ↩︎
-
(Spann, J. F., Bove, A. A., Natarajan, G. & Kreulen, T. Ventricular performance, pump function and compensatory mechanisms in patients with aortic stenosis. Circulation 62, 576–582 (1980)) ↩︎
-
(Davies, C. H. et al. Reduced Contraction and Altered Frequency Response of Isolated Ventricular Myocytes From Patients With Heart Failure. Circulation 92, 2540–2549 (1995)) ↩︎
-
(Beuckelmann, D. J., Näbauer, M. & Erdmann, E. Intracellular calcium handling in isolated ventricular myocytes from patients with terminal heart failure. Circulation 85, 1046–1055 (1992)) ↩︎
-
(Bers, D. M., Eisner, D. A. & Valdivia, H. H. Sarcoplasmic Reticulum Ca2+ and Heart Failure Roles of Diastolic Leak and Ca2+ Transport. Circ. Res. 93, 487–490 (2003)) ↩︎
-
(Margulies, K. B. & Houser, S. R. in Heart Fail. Companion Braunwalds Heart Dis. Second Ed. (Mann, D. L.) 32–47 (W.B. Saunders, 2011). at http://www.sciencedirect.com/science/article/pii/B9781416058953100038) ↩︎
-
(Bousette, N. et al. Large-scale characterization and analysis of the murine cardiac proteome. J. Proteome Res. 8, 1887–1901 (2009)) ↩︎
-
(Shannon, T. R., Wang, F., Puglisi, J., Weber, C. & Bers, D. M. A mathematical treatment of integrated) ↩︎