Dual-use note. This post describes a defensive screening layer for synthesized DNA. It deliberately reports aggregate metrics on a curated, public-source benchmark and omits operational details that could inform evasion (per-mutation-rate ladders, short-read false-alarm tables, dataset assembly recipes, attack pipelines). Code and processed data are available on request to vetted researchers. The findings here are complementary to alignment- and HMM-based policy screens (e.g. IBBIS Common Mechanism); they are not a substitute for them.
DNA-synthesis screening today asks a narrow question: does this order look like something on a known-toxin list? Letter-by-letter alignment against curated databases catches the easy attacks and the honest-mistake orders, but it has a known failure mode: change enough nucleotides to escape the lookup while keeping the protein’s function, and the screen falls silent (Wittmann & Olsen et al., 2024).
Function-level screening (does this sequence look like it does what a toxin does?) is what large biological sequence models are good at. This post summarizes a homology-aware benchmark of two such models, ESM-2 (protein) and Evo2 7B (DNA), framed as a complementary layer behind alignment-based triage.
Table of Contents
- The screening gap
- Benchmark
- What the embeddings see
- Headline result
- Disguise resilience, qualitatively
- Where this fits in a screening stack
- Limits and responsible use
The screening gap
A modern DNA synthesis order goes through a tiered review. Letter-matching against curated hazard databases is fast, precise, and cheap, and it is what most public-facing screens use. The structural problem is that protein function is not preserved by surface DNA similarity: silent codon changes can rewrite the DNA letters of a toxin while leaving the protein, and its harmful activity, intact. The reverse is also true: adversaries (or evolution) can mutate amino acids enough to break alignment while folding to the same toxic 3D shape.
A function-aware screen would read past surface spelling. The question is whether learned representations from large biological models, which were never trained on a “toxin / not-toxin” label, encode enough of “what the protein does” to act as that second layer.
Benchmark
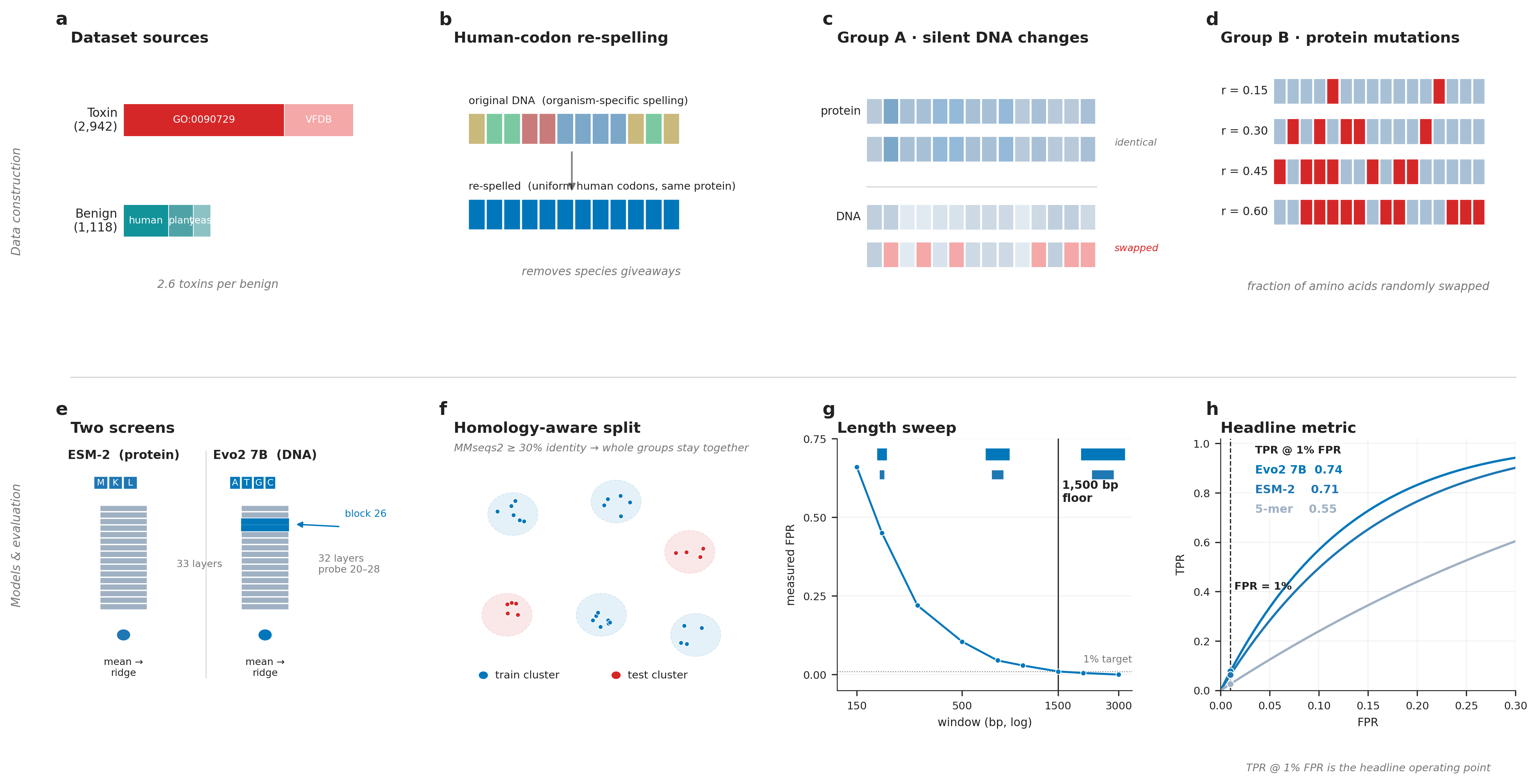
We assemble a labeled, homology-aware benchmark of toxin and benign coding sequences:
- Toxins drawn from the Gene Ontology toxin activity category and the Virulence Factor Database.
- Benigns drawn from human-secreted, plant, and yeast proteins, none of which appear in the toxin catalogs.
- Total: 2,942 toxins / 1,118 benigns after pairing DNA and protein records.
Two design choices matter:
- Human-codon re-spelling. Every coding sequence is rewritten with uniform human codons before any DNA model sees it. This forces any DNA-side signal to come from biology, not species-of-origin spelling.
- Homology-aware split. We group sequences by MMseqs2 at 30% protein identity and keep entire groups together in either training or testing, never split across both. Without this guard, close homologs leak between folds and inflate every metric.
The headline metric is TPR@1%: the fraction of toxins caught when the alarm threshold is set so that 1% of benign sequences trigger an alert. This is the operating point most relevant to high-throughput synthesis-order review.
What the embeddings see
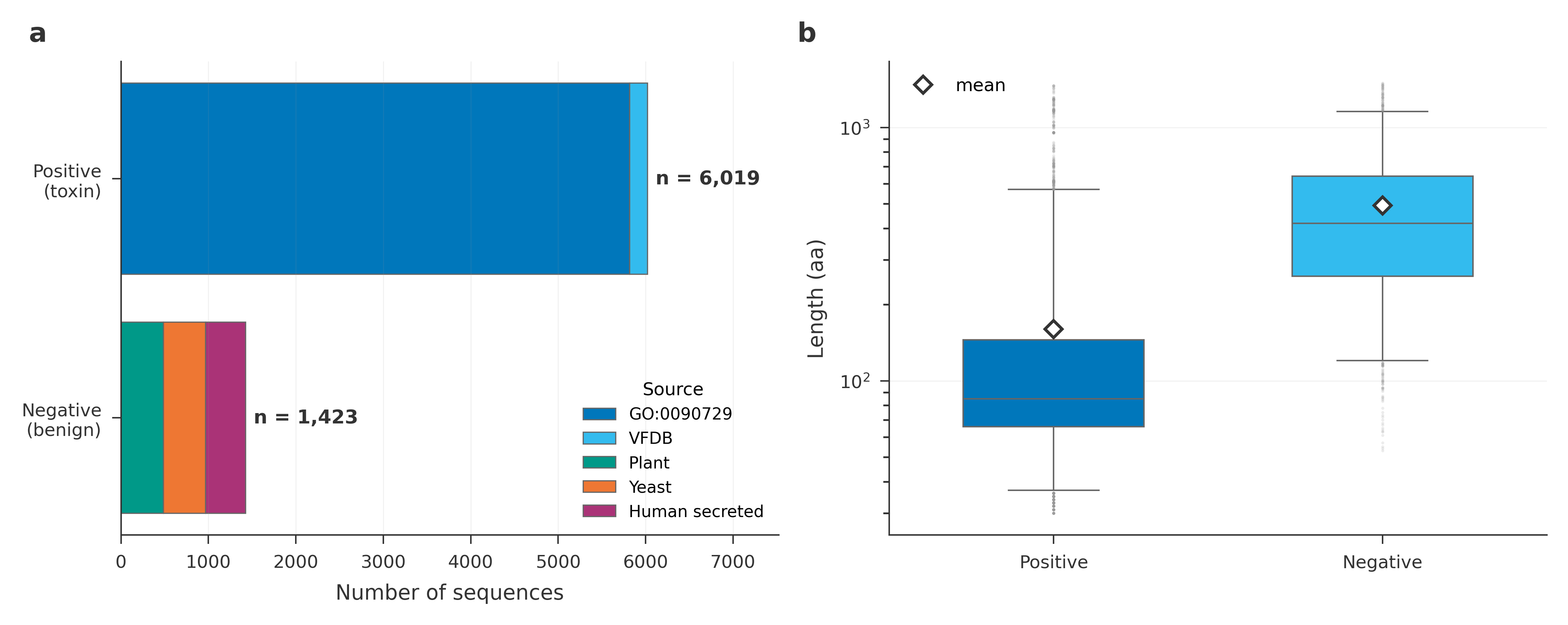
Before training any classifier, a UMAP projection of mean-pooled embeddings already separates the signal:
ESM-2’s protein embeddings cleanly split toxin from benign. The DNA-side picture is more layered: small DNA models leave the classes intermingled, while Evo2 7B forms two structured clusters that separate further at intermediate layers than at the final layer. The takeaway is that the DNA-side functional signal is not in the model’s surface output; it lives in the upper-middle of the stack, consistent with deeper layers encoding what the protein does rather than its raw letter frequencies.
We use this as motivation for a layer probe: train a linear classifier on each Evo2 block in a chosen window and pick the layer with the strongest function signal as the deployed DNA screen.
Headline result
Given returned positive set $R$ and ground truth $G$, define the catch-rate at a fixed false-alarm rate $\alpha$:
$$ \text{TPR}@\alpha ;=; \Pr!\left[\hat{y}=1 \mid y=1\right];;\text{s.t.};;\Pr!\left[\hat{y}=1 \mid y=0\right] = \alpha. $$
A simple z-score average of the ESM-2 and Evo2 decision streams,
$$ s_{\text{ens}}(x) ;=; \tfrac{1}{2}!\left(\frac{s_{\text{ESM}}(x) - \mu_{\text{ESM}}}{\sigma_{\text{ESM}}} + \frac{s_{\text{Evo2}}(x) - \mu_{\text{Evo2}}}{\sigma_{\text{Evo2}}}\right), $$
beats either model alone:
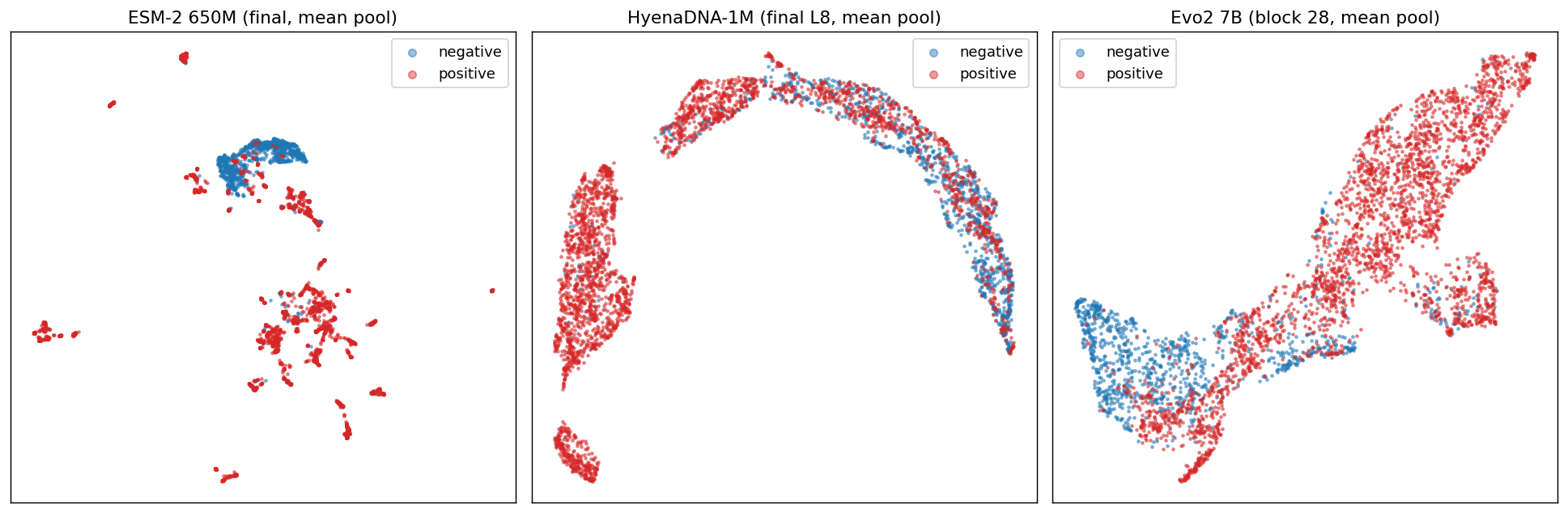
| Screen | AUROC | TPR@1% |
|---|---|---|
| ESM-2 + Evo2 ensemble (z-avg) | 0.993 | 0.858 |
| Evo2 (best probed layer) | 0.990 | 0.722 |
| ESM-2 | 0.985 | 0.705 |
| 5-letter-pattern baseline | 0.869 | 0.548 |
Two observations.
The ensemble is doing real work, not riding on one model. The two screens disagree often enough on hard cases that averaging their standardized scores recovers an extra ~13 percentage points of toxins at the 1% target with no retraining. Most of the lift shows up at stricter operating points, where the marginal toxin matters most.
Function signal beats letter-frequency signal by a wide margin. The 5-letter-pattern baseline, which sees only DNA composition, sits 30 points below the learned screens. This is the part of the gap a function-aware layer is designed to close.
Disguise resilience, qualitatively
Two forms of disguise stress the screen in different ways:
- Silent DNA changes rewrite the nucleotide spelling of a toxin while leaving the protein identical. This is the easiest way to defeat letter-matching alone.
- Protein-level mutation randomly substitutes amino acids; with enough substitutions, surface alignment fails even at the protein level.
The qualitative finding (and the part safe to share publicly) is that the protein screen’s catch rate is flat across silent DNA changes and degrades only gently as amino-acid substitutions accumulate, while a letter-frequency baseline degrades sharply on the same inputs.
The mechanism is the one the UMAP picture already hinted at: the protein screen tracks what the protein does and survives small changes in its sequence; the letter-frequency screen tracks DNA spelling and breaks down when those letters are disturbed.
We deliberately do not publish the per-mutation-rate ladder, the BLAST-evasion-rate breakdown, or the short-read false-alarm tables in this post. They are operationally useful for stress-testing real screening pipelines and we are happy to share them with vetted researchers and screening providers; they are not the right primitives for a public blog.
Where this fits in a screening stack
The right way to read these numbers is complementary, not competing.
╭───────────────────────────╮ ╭────────────────────────────╮
│ Tier 1 │ │ Tier 2 │
│ alignment + HMM │ ──▶ │ function-aware │
│ (e.g. Common Mechanism) │ │ embedding ensemble │
│ high precision on │ │ generalises to families │
│ known regulated families │ │ outside the curated list │
╰─────────────┬─────────────╯ ╰────────────┬───────────────╯
│ │
▼ ▼
flag for review flag for review
│ │
└──────────────┬─────────────────┘
▼
human-in-the-loop triage
A practitioner-oriented take:
- Pair a letter-matching filter with a protein-level screen whenever an attacker could mutate the toxin while keeping its 3D shape.
- Re-tune the alarm threshold for each deployment setting; do not reuse a paper’s false-alarm rate on a 150-bp short-read pipeline.
- For the highest-stakes review tier, deploy the z-scored ESM-2 + Evo2 ensemble; recovers ~15 percentage points more toxins at 1% FPR than single models.
- Reliable behavior in this benchmark required DNA fragments at least on the order of ~1.5 kb; short-read contexts (metagenomics, wastewater) need different operating points and follow-up confirmation.
Limits and responsible use
The honest caveats:
- Random mutations are not structure-guided escape. They stress the screen but do not target alignment blind spots specifically. Any regulator-facing claim should re-run the evasion test with a structure-guided protocol on an independent, unseen set.
- Threshold transfer is fragile on small benign holdouts. A 1% target calibrated on ~150 holdout benigns lands at a different effective FPR on the test set. A larger benign holdout closes this directly.
- Ground truth is curated, not exhaustive. Toxins outside GO:0090729 / VFDB are not labeled positive even when they may be biologically harmful, and the converse holds for the benign side.
- One benchmark, one screening tier. Real synthesis-order review is layered, regulated, and includes human triage. A learned screen is one input, not the decision.
On the dual-use side, two principles drove what is and is not in this post: publish enough to convince defenders the gap exists and that learned models help close it, and keep operational specifics (per-mutation tables, short-read floors, dataset assembly recipes, code) gated behind request access. If you are at a screening provider, an academic lab working on biosecurity, or a public-health authority, get in touch.
Citation
Amanfu, Robert. "Beyond Sequence Similarity: Function-Aware Screening of Synthesized DNA."
Robert's Bytes (May 2026). https://pubs.onrender.com/posts/beyond_sequence_similarity/
References
- Wittmann, B. J. et al. (2024). Toward AI-Resilient Screening of Nucleic Acid Synthesis Orders. bioRxiv. doi:10.1101/2024.12.02.626439
- Wheeler, N. E. et al. (2024). Developing a Common Global Baseline for Nucleic Acid Synthesis Screening. Applied Biosafety 29(2):71–78. doi:10.1089/apb.2023.0034
- Lin, Z. et al. (2023). Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379:1123–1130. [ESM-2]
- Arc Institute / Stanford / UC Berkeley (2025). Evo2 7B model card. huggingface.co/arcinstitute/evo2_7b
- Tayouri, S. et al. (2025). Defending Synthetic DNA Orders Against Splitting-Based Obfuscation. bioRxiv. doi:10.1101/2025.03.12.642526
- Steinegger, M. & Söding, J. (2017). MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nature Biotechnology 35:1026–1028. doi:10.1038/nbt.3988
- IBBIS (2024–2026). Common Mechanism for DNA Synthesis Screening (
commec). github.com/ibbis-bio/common-mechanism